EU AI Act 디지털 옴니버스 합의: 한국 SaMD 기업이 2028년까지 해야 할 일
2026년 5월 EU가 합의한 디지털 옴니버스는 의료기기 AI 고위험 의무를 2028년 8월로 연기했다. 하지만 Notified Body 대기열과 이중 적합성 평가 구조를 고려하면, 지금 준비를 멈추는 건 가장 위험한 선택이다.
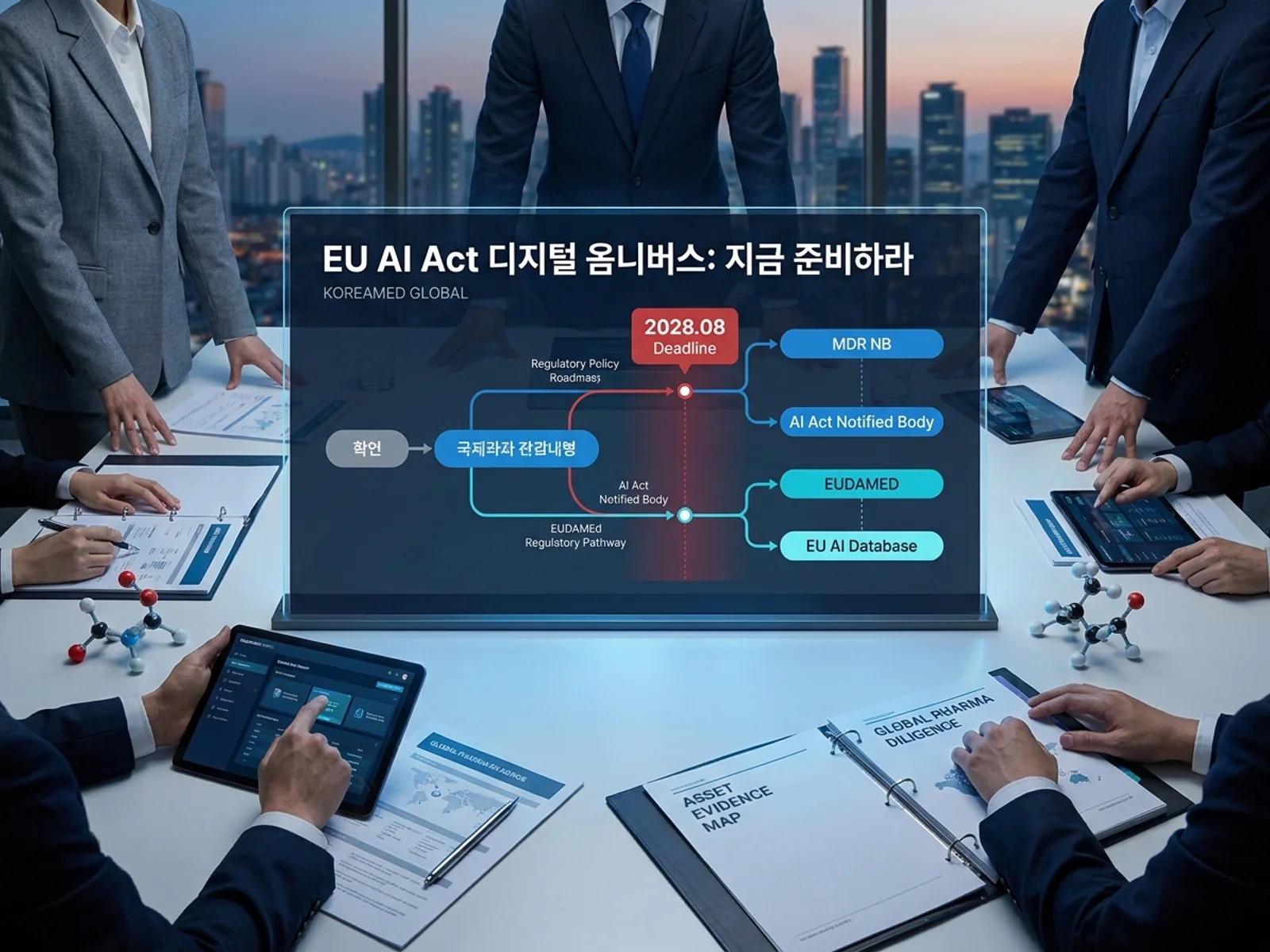
왜 지금 이 이슈를 봐야 하나
2026년 5월 7일, EU 이사회와 유럽의회는 AI Act 개정안인 Digital Omnibus on AI에 잠정 합의했다. 의료기기 업계에 가장 중요한 변화는 하나다. MDR·IVDR 적용을 받는 의료기기에 내장된 AI 시스템(Article 6(1), Annex I)의 고위험 AI 의무 적용일이 2027년 8월에서 2028년 8월 2일로 1년 연기된다.
한국 SaMD·디지털헬스 기업 입장에서 이것은 "준비할 시간이 늘었다"가 아니라 "준비가 더 늦어지면 치명적"이라는 의미다. Notified Body(NB) 대기열이 현재 18~24개월이고, AI Act 적합성 평가를 담당할 NB 지정은 2026년 중반까지도 소수에 불과하다. 2028년 8월 deadline은 NB 대기열의 같은 쪽에 있다.
핵심 타임라인 비교
| 항목 | 기존 deadline | 변경 후 | 비고 |
|---|---|---|---|
| Annex III 독립형 고위험 AI | 2026년 8월 2일 | 2027년 12월 2일 | 16개월 연기 |
| Annex I 제품 내장형 고위험 AI(의료기기) | 2027년 8월 2일 | 2028년 8월 2일 | 12개월 연기 |
| Article 50 투명성 의무(워터마크, 출처 표시) | 2026년 8월 2일 | 그대로 | 영향 없음 |
| AI 생성 CSAM·디핀센셜 이미지 금지 | 신규 | 2026년 12월 2일 | 새로 추가 |
| 정식 채택·관보 게재 | — | 2026년 8월 2일 이전 예상 | 이전까지 구법이 현행 |
출처: Bird & Bird, "Digital Omnibus on AI Provisional Agreement Reached at the May Trilogue" (2026년 5월); Gibson Dunn, "EU AI Act Omnibus Agreement" (2026년 5월); European Parliament press release (2026년 5월 7일).
한국 SaMD 기업이 당장 확인해야 할 것
1. 내 제품이 고위험 AI인가
EU AI Act Article 6(1)에 따르면, MDR Class IIa 이상 또는 IVDR Class B 이상 의료기기에 AI 컴포넌트가 포함되어 있고 Notified Body 적합성 평가가 필요한 경우, 자동으로 고위험 AI로 분류된다.
- MDR Class I 자가인증 → 고위험 AI 아님(단, 멸균·측정·재사용 수술기구 서브카테고리는 NB 관여 가능하므로 재확인 필요)
- IVDR Class A 자가인증 → 대부분 고위험 아님
- 그 외 Class IIa, IIb, III → 고위험 AI 확정
MDCG 2025-6(2025년 6월 발표)이 "Medical Device Artificial Intelligence(MDAI)" 정의를 명확히 했다. 자사 제품의 intended use에 AI가 포함되어 있는지, 아니면 단순 rule-based 소프트웨어인지 먼저 구분하라.
2. NB에 AI Act 지정 상태를 물어라
이것이 단기적으로 가장 중요한 액션 아이템이다. 고위험 AI 의료기기의 적합성 평가는 기존 MDR/IVDR NB가 AI Act까지 담당하는 구조다. 별도의 NB가 추가되는 게 아니다.
2026년 중반 기준, AI Act 지정을 완료한 NB는 극소수다. 한국 기업이 MDR 인증을 받은 NB가 AI Act 심사 역량을 갖추고 있는지 지금 확인해야 한다. 답이 "아직"이면, 그것이 2028 deadline에서 가장 큰 스케줄링 리스크다.
실무 팁: NB에 세 가지를 물어라.
- AI Act 지정 신청 상태
- 지정 예상 scope(제품 유형 포함)
- 자사 기기가 그 scope에 포함되는지
3. 이중 등록 준비: EUDAMED ≠ EU AI Database
자주 나오는 오해가 있다. "EUDAMED에 등록되어 있으면 AI Act도 자동 충족된다." 그렇지 않다.
EUDAMED은 MDR/IVDR 등록 시스템이다. AI Act는 별도의 EU AI Database에 고위험 AI 시스템을 등록해야 한다. 두 개의 등록 체계를 독립적으로 준비해야 한다. 2028년 8월 이전에 EU AI Database가 실제로 가동될지도 불확실하며, Council 버전은 데이터베이스 실제 가용성을 deadline 조건으로 명시하기도 했다.
Articles 9–17: 8개 기술 의무, 연기된 건 아무것도 없다
Omnibus가 바꾼 건 적용 시점뿐이다. 의무 내용 자체는 그대로다.
| Article | 의무 | MDR과의 관계 | 한국 기업이 신경 써야 할 포인트 |
|---|---|---|---|
| 9 | AI 위험관리시스템 | ISO 14971과 병행, 대체 불가 | 환자 안전 + 기본권·공정성 리스크까지 포함 |
| 10 | 데이터 거버넌스 | 임상평가와 겹치지만 AI 특화 요건 추가 | 학습·검증·테스트 데이터의 출처·대표성·편향 검증 |
| 11 | 기술문서 | MDR Annex II/III와 중복 + AI 특화 첨부 | 학습 방법론, 성능 지표, 한계, 오용 가능성 명시 |
| 12 | 기록 관리(자동 로깅) | MDR에 없는 신규 요건 | 로깅은 아키텍처 결정이라 6~18개월 소요 가능 |
| 13 | 투명성·사용자 정보 | IFU와 연계 | AI 작동 방식, 한계, 성능에 대한 정보 제공 |
| 14 | 인적 감독 | 의사 결정권 보장 설계 | AI 출력에 대한 의사 override 메커니즘 |
| 15 | 정확성·견고성·사이버보안 | MDR 사이버보안과 겹침 | 모델 드리프트 모니터링, 적대적 공격 방어 |
| 16 | 고위험 AI 제공자 의무 | Section 2 요건 준수, 적합성 평가, CE 마킹, EU DB 등록 | 제공자(provider)로서 전 lifecycle 의무 |
| 17 | 품질경영시스템 | 기존 ISO 13485 QMS에 AI 절차 추가 | 별도 QMS 만들지 말고 기존 체계에 통합 |
출처: EU AI Act Regulation 2024/1689, Chapter III, Section 2; RegAffairs Hub, "EU AI Act for Medical Device Software" (2026년 5월).
한국 AI 기본법과 EU AI Act의 병행 대응
2026년 1월 22일 발효된 한국 AI 기본법도 의료 AI를 "고영향 AI"로 분류한다. 두 법제의 요구사항이 상당 부분 겹친다.
| 공통 요소 | 한국 AI 기본법 | EU AI Act |
|---|---|---|
| 위험관리계획 | 필수 | 필수(Article 9) |
| 인적 감독 | 필수 | 필수(Article 14) |
| 설명 가능성 | 한국어 설명 자료 제공 | 투명성 의무(Article 13) |
| 데이터 거버넌스 | — | 상세 요건(Article 10) |
| 적합성 평가 | DMPA 인허가 절차와 병행 | NB를 통한 이중 적합성 평가 |
한국 내수와 EU 수출을 동시에 하는 기업이라면, 두 규제의 문서화 요건을 병렬로 준비하는 것이 효율적이다. 같은 위험관리 문서를 한국어와 영문으로 각각 작성하는 대신, 하나의 마스터 문서에서 양쪽 요건을 충족하는 구조로 설계하라.
2026년 하반기 실행 계획
Q3 2026: 분류·갭 분석
- 자사 제품이 고위험 AI인지 확정
- 기존 MDR 기술문서와 AI Act Articles 9–17 요건 갭 분석
- NB에 AI Act 지정 상태 확인
Q4 2026: 데이터 거버넌스 베이스라인
- 학습·검증·테스트 데이터의 출처, 범위, 한계 문서화
- 편향 검사(bias examination) 단계 추가
- 이 작업이 보통 가장 오래 걸린다 — 지금 시작하라
Q1 2027: QMS 확장
- AI 특화 절차(데이터 거버넌스, 모델 변경 관리, AI 인시던트 보고, 사후 모니터링)를 기존 ISO 13485 QMS에 추가
- 별도 QMS를 만들지 말고 기존 체계에 통합
Q2 2027: NB와의 사전 조율
- AI Act 적합성 평가가 MDR 감사와 어떻게 통합되는지 NB와 합의
- 기술문서 AI Act 보충 분석 초안 제출
Q3–Q4 2027: 최종 문서 제출
- EU AI Database 등록 준비
- MDR Annex II/III + AI Act Annex IV 통합 기술문서 완성
실무에서 자주 묻는 질문
"이미 MDR CE 마크를 받은 제품도 해당되나"
2028년 8월 2일 이전에 이미 시장에 출시된 AI 의료기기는, 설계에 "중대한 변경(significant change)"을 하지 않는 한 고위험 AI 의무가 소급 적용되지 않는다(MDCG 2025-6 Q31). 단, "중대한 변경"의 정의가 아직 불명확하다. 모델 업데이트, 새로운 임상 적응증 추가, 알고리즘 아키텍처 변경은 중대한 변경으로 간주될 가능성이 높다.
"한국에서만 판매하면 EU AI Act와 무관한가"
그렇다. EU 시장에 출시하지 않으면 AI Act 의무가 없다. 하지만 한국 AI 기본법의 고영향 AI 의무가 별도로 적용된다. 두 시장을 동시에 공략하는 기업이라면 이중 준비가 필요하다.
"Omnibus가 추가 연기될 수 있나"
가능하다. Council 버전은 조화 표준(harmonised standards)과 EU AI Database의 실제 가용성을 deadline 조건으로 명시했다. 하지만 기다리는 전략은 위험하다. NB 대기열은 줄어들지 않고, 아키텍처 결정(로깅, 인적 감독, 데이터 거버넌스)은 6~18개월이 걸린다.
결론: 연기는 준비 시간이지, 준비 면제가 아니다
2028년 8월 deadline은 NB 대기열 관점에서 여전히 같은 쪽에 있다. 지금 준비를 멈추면 2027년에 뒤에서 시작하게 된다. 한국 SaMD 기업이 취해야 할 태도는 명확하다.
- 지금 분류하고 갭 분석하라 — 제품이 고위험 AI인지, 어떤 Article이 빠져 있는지
- NB에 물어라 — AI Act 지정 상태, scope, 자사 제품 포함 여부
- 한국 AI 기본법과 병행 준비하라 — 문서화 요건이 겹치는 부분을 하나의 프로세스로 관리
- 아키텍처 결정을 올해 안에 내려라 — 로깅, 인적 감독, 데이터 거버넌스는 나중에 추가하기 어렵다


